Greg informed me that the Perl scripts are there just for reference and he had to make many changes to it for the paper. The scripts he used for the paper are gone because it was stored on scratch space. So I decided to dump the Perl scripts and start from scratch. According to the paper, "CG and CHG DMRs were required to have a minimum of 3 symmetrical CG or CHG sites, at least 2X coverage, and a minimum methylation difference between two genotypes of 60%. CHH sequence context DMRs were required to have a minimum of 6 asymmetrical CHH sites, a minimum of 2X coverage, and one genotype with <5% methylation and the other genotype with >25% methylation." Luckily for me, the tab file output from the zed-align has all of these information. The tab file contains the number of CG, number of Methylated CG, CG ratio, etc. With these information, I can just parse the file line by line to get their methylation. Then, I can compare it with the methylation of another tab file to determine if the region is differentally methylated. After a while, I completed the script. To my surprise, my script uses only 3.2 MB of ram and finish parsing through 2 1.5 GB tab files in about 7 minutes.
|
One missing component of the zed-align pipeline that is in the paper "Natural Variation of DNA Methylation in Maize" is the comparisons between two genomes to locate the differentially methylated regions. In the paper, they used a series of perl scripts for alignments and comparisons. Despite the different inputs and outputs of the perl scripts, the zed-align pipeline performs similar functions, and I'm trying to implement the comparison functionality. My assignment is to convert the perl scripts into python scripts so that it can be implemented into the zed-align pipeline. My current approach is to convert each of the perl script into python script, so that is is in a language that I am familiar with. Then, tweak the script to accept the inputs from the zed-align pipeline. However, I am getting a lot of errors from the perl scripts and currently investigating the cause of the errors. 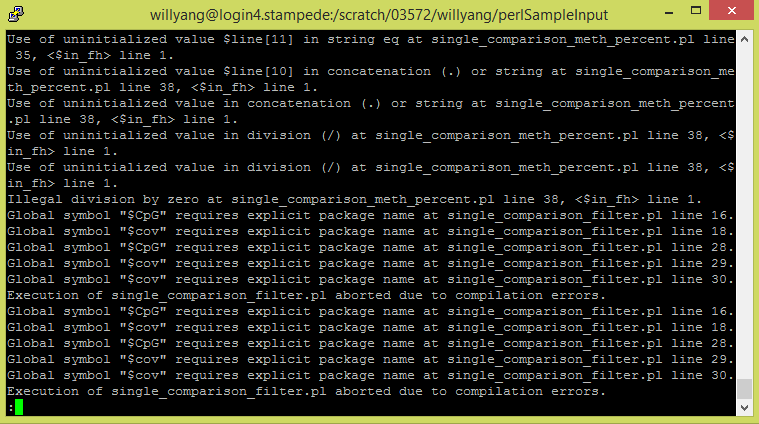 Greg informed me that we have a group meet at the end of this week. Since most of the people in the group are not informed on the details of his pipeline, my assignment for the week is to study the pipeline and present the details of his pipeline to the group. The ZED-align pipe takes 2 fastq files and a fq file as input. The fastq file contains 100 base pairs of DNA sequences that are bisullfite sequenced. Bisulfite sequencing is the process where all the non-methylated cytosine bases into thymine. So the DNA began in the lab and was randomly spliced. Then, the DNA segments was bisulfite sequenced and read by an interval of 100 base pairs to produce the fastq file. The fq file on the other hand contains the genomic data, which will be used to match the base pairs on the fastq file. The ZED-align first find the location of the 100 base pairs on the specified chromosome. However, due to the duplicates, this might not be successful for every 100 base pairs. For example, the chromosome might contain multiple AATTCT. In addition, the chromosome might also contain AACCCC, etc. So it is unclear whether those thymines are actually thymines or converted cytosines. As a result, not all base pairs are aligned. Despite this, most alignments are successful as the probability for duplicate 100 base pairs is quite low. After the alignment, the pipeline will calculate the methylation of the 100 base pairs and output three different methylation tiles: CG, CHG, CHH. The three tiles shows the amount of a particular methylation that is in the 100 base pairs. C stands for cytosine, G stands for Guanine, and H stands for anything buy Guanine. So for example, CCG has CG methylation and CHG methylation. 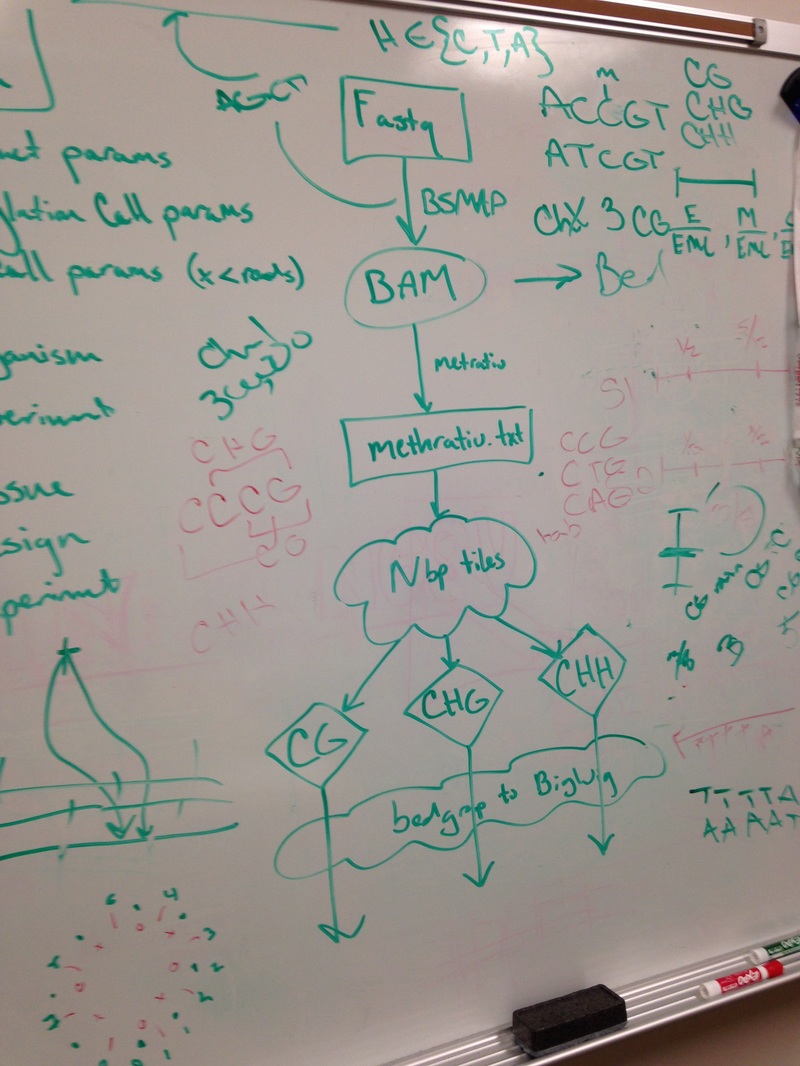 Greg introduced me to a pipeline that he has been working on called ZED-align, an alignment and methylation calling pipeline for Zea Epigenomics Database. Greg gave me the 5geno sample, which contains 5 pairs of reads, each with the size about 40GB, to run on his pipeline. Since Greg estimated that it would take 8 hours to run each pair of reads and each pair of reads are distinct calculations, running all 5 pairs of reads in a sequential manner would take about 40 hours, which is way too inefficient. As a result, Greg gave me the task to parallelize the run using the module launcher. After digging around the documentations for launcher, I learned that the module parallelizes batch jobs by running each line of batch commands on different nodes. Therefore, I created a simple batch script that locate all the pairs of reads (in fq format) and echo each command on a new line. To my surprise, the pipe finished the 5 pairs of reads in about 7 hours.
Last summer, I visited UT Austin on my way to Houston. The visit was very short, and I only took several pictures before departing. I would had never guess that I would be back this summer, not only to visit, but to stay for 2 months. One thing that I failed to notice during the brief visit is the sheer size of the campus. The size of UT Austin campus still amazes me to this day.
I was very nervous about meeting other ICERT researchers in the REU program. This is because I did not communicate with them in groupme prior due to a mistake on my phone number, and I was afraid that I missed out on a lot of information. However, as it turns out, not much has been said on groupme and everyone was as confused as I am. For the weekend, we went to lunch and dinner together. The transition for me was quick and smooth. Monday was my first day at TACC. One of the first thing I saw when I arrived to TACC was their supercomputers. This was the first time I saw a supercomputer and I was very fascinated by the sheer size and power these machines possessed. For the next 3 days, I went through many trainings. Although they are very long, I learned a great deal from them. On Thursday, I meet my mentor, Dr Matthew Vaughn, for the first time and discovered the details of my REU project. I also learned that I will be mostly working with his assistant, Greg Zynda. I am very excited about my project and can't wait to start working on it. |
 RSS Feed
RSS Feed
