Greg informed me that we have a group meet at the end of this week. Since most of the people in the group are not informed on the details of his pipeline, my assignment for the week is to study the pipeline and present the details of his pipeline to the group.
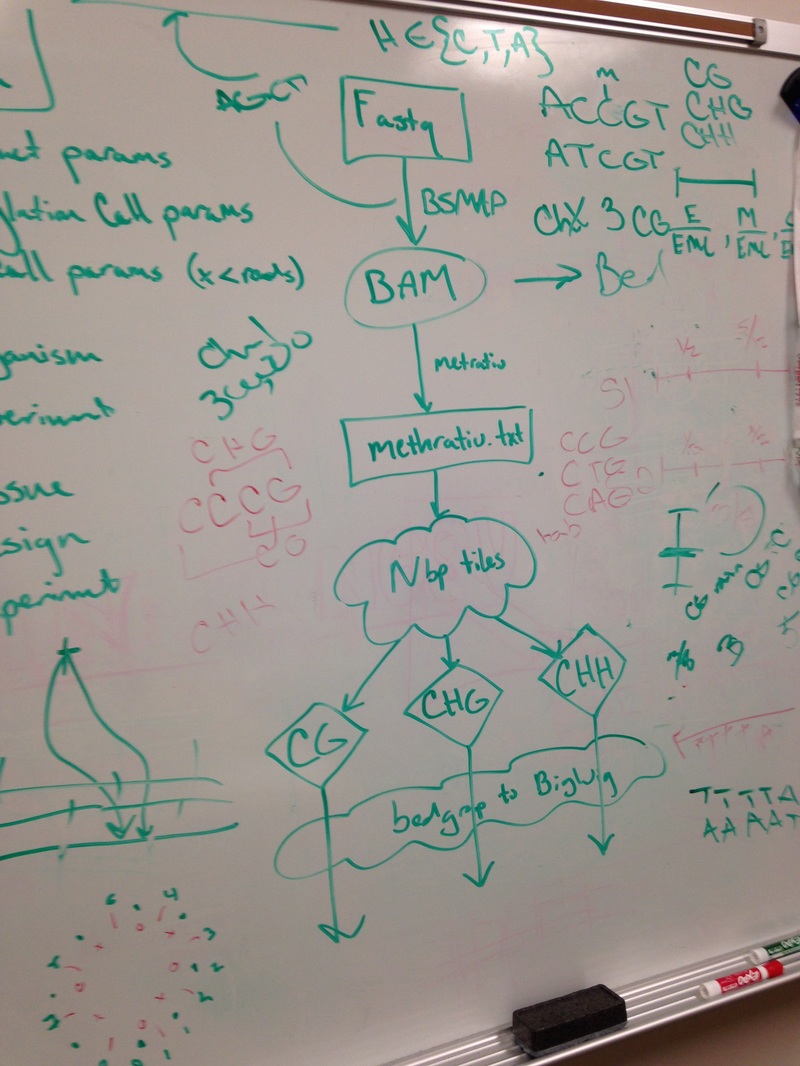
The ZED-align pipe takes 2 fastq files and a fq file as input. The fastq file contains 100 base pairs of DNA sequences that are bisullfite sequenced. Bisulfite sequencing is the process where all the non-methylated cytosine bases into thymine. So the DNA began in the lab and was randomly spliced. Then, the DNA segments was bisulfite sequenced and read by an interval of 100 base pairs to produce the fastq file. The fq file on the other hand contains the genomic data, which will be used to match the base pairs on the fastq file.
The ZED-align first find the location of the 100 base pairs on the specified chromosome. However, due to the duplicates, this might not be successful for every 100 base pairs. For example, the chromosome might contain multiple AATTCT. In addition, the chromosome might also contain AACCCC, etc. So it is unclear whether those thymines are actually thymines or converted cytosines. As a result, not all base pairs are aligned. Despite this, most alignments are successful as the probability for duplicate 100 base pairs is quite low.
After the alignment, the pipeline will calculate the methylation of the 100 base pairs and output three different methylation tiles: CG, CHG, CHH. The three tiles shows the amount of a particular methylation that is in the 100 base pairs. C stands for cytosine, G stands for Guanine, and H stands for anything buy Guanine. So for example, CCG has CG methylation and CHG methylation.
The ZED-align pipe takes 2 fastq files and a fq file as input. The fastq file contains 100 base pairs of DNA sequences that are bisullfite sequenced. Bisulfite sequencing is the process where all the non-methylated cytosine bases into thymine. So the DNA began in the lab and was randomly spliced. Then, the DNA segments was bisulfite sequenced and read by an interval of 100 base pairs to produce the fastq file. The fq file on the other hand contains the genomic data, which will be used to match the base pairs on the fastq file.
The ZED-align first find the location of the 100 base pairs on the specified chromosome. However, due to the duplicates, this might not be successful for every 100 base pairs. For example, the chromosome might contain multiple AATTCT. In addition, the chromosome might also contain AACCCC, etc. So it is unclear whether those thymines are actually thymines or converted cytosines. As a result, not all base pairs are aligned. Despite this, most alignments are successful as the probability for duplicate 100 base pairs is quite low.
After the alignment, the pipeline will calculate the methylation of the 100 base pairs and output three different methylation tiles: CG, CHG, CHH. The three tiles shows the amount of a particular methylation that is in the 100 base pairs. C stands for cytosine, G stands for Guanine, and H stands for anything buy Guanine. So for example, CCG has CG methylation and CHG methylation.
 RSS Feed
RSS Feed
